Int J Med Sci, 01/01/2026

Introduction
Stroke remains one of the leading causes of death and long-term disability worldwide, with an increasing disease burden over the past decade. Despite advances in early interventional therapies such as tissue plasminogen activator (tPA) and mechanical thrombectomy, treatment options remain limited, and many patients continue to suffer from persistent neurological sequelae. Stem cell–based therapies, with the potential to promote tissue regeneration, repair damaged neurons, and restore lost functions through stimulation of neurogenesis and angiogenesis, are emerging as a promising approach in stroke treatment. Recent studies indicate that stem cells can migrate to injured brain regions, differentiate into neural cells, and integrate into existing neural networks, thereby potentially improving functional recovery.
Mesenchymal stem cells (MSCs), one of the most commonly used stem cell types, have demonstrated potential in post-stroke recovery in preclinical studies. Among MSCs, umbilical cord–derived mesenchymal stem cells (UCMSCs) have attracted increasing attention due to their ease of isolation, low immunogenicity, and strong regenerative capacity. UCMSCs can home to sites of injury via chemokine-mediated targeting mechanisms and promote tissue repair through paracrine signaling. These cells exert therapeutic effects by modulating inflammatory responses, enhancing angiogenesis, and promoting neurogenesis, thereby contributing to functional recovery after stroke. Moreover, UCMSCs have a favorable safety profile, making them a promising candidate for clinical applications.
Although preclinical results have been highly encouraging, there are still few clinical trials investigating the therapeutic application of UCMSCs in acute ischemic stroke (AIS). The heterogeneity of stroke pathogenesis, differences in treatment protocols, and challenges in standardizing cell-based therapies have hindered progress in this field. To address the unmet therapeutic need, we initiated a phase I, open-label, dose-escalation, first-in-human study to evaluate the safety and tolerability of UMC119-06, a UCMSC product, in patients with AIS.
The primary objective is to assess the safety, tolerability, and maximum feasible dose (MFD) of UMC119-06. Secondary objectives include evaluation of long-term safety and preliminary clinical efficacy following a single intravenous infusion of UMC119-06. In addition, the study investigates the effects of UMC119-06 on stroke-related biomarkers, which may provide further insights into its mechanism of action and potential therapeutic benefits. Ultimately, this study aims to provide foundational data to guide subsequent investigations of UCMSC-based therapies in the treatment of AIS.
Methods
Protocol approvals, registration, and informed patient consent
The UMC119-06 trial protocol (ClinicalTrials.gov identifier: NCT04097652) was approved by the Taiwan Food and Drug Administration (No. 1086027359) and the Institutional Review Board of Taipei Medical University – Shuang Ho Hospital (N201904089). The study was conducted in accordance with the Declaration of Helsinki, and informed consent was obtained from all participants or their legally authorized representatives following a thorough explanation.
Trial design and oversight
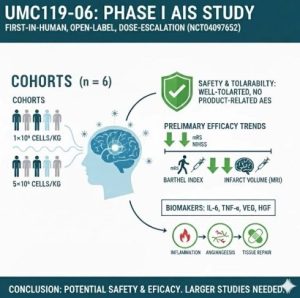
This was a phase I, open-label, dose-escalation clinical trial designed to evaluate the safety, tolerability, and maximum feasible dose (MFD) of UMC119-06 in patients with acute ischemic stroke (AIS). The study employed a standard 3+3 dose-escalation design, in which each dose level included initial sentinel dosing to assess safety before cohort expansion. Escalation to the next dose level was permitted when 0 of 3 patients or ≤1 of 6 patients experienced a dose-limiting toxicity (DLT); conversely, enrollment was halted if ≥2 patients experienced DLTs. The trial design schema is shown in Figure 1.

Figure 1. Study Diagram for Trial Design and Oversight.
UMC119-06 is a human umbilical cord–derived mesenchymal stem cell (UCMSC) product manufactured in a Good Tissue Practice (GTP)–compliant laboratory for cell therapy (Reon Biotech, Taiwan). The product was formulated in 0.9% NaCl solution supplemented with 2% clinical-grade human serum albumin, with a cell density ranging from 3.3×10⁵ to 2.3×10⁶ cells/mL.
Dose selection for human administration was based on preclinical animal studies using three different dose levels, in which no significant safety concerns were observed. To translate these findings into an appropriate clinical dose, the investigators applied principles of interspecies dose conversion using allometric scaling, taking into account differences in body weight, cell distribution, as well as the viability and biological activity of the final product. In this study, eligible patients were assigned to either dose level 1 (1×10⁶ cells/kg) or dose level 2 (5×10⁶ cells/kg) and received a single intravenous infusion of UMC119-06.
Participants
Eligible patients were aged between 20 and 80 years, with a stroke onset within 48 to 168 hours before the start of treatment, and the stroke was caused by large-artery atherosclerosis or cardioembolism. Patients were required to have a pre-treatment modified Rankin Scale (mRS) score of 0 or 1, National Institutes of Health Stroke Scale (NIHSS) scores between 5 and 20, and no increase of ≥4 points in NIHSS scores from baseline. The presence of a hemispheric cortical infarct was confirmed by brain magnetic resonance imaging (MRI), with a lesion size of less than 100 mL on diffusion-weighted imaging. Exclusion criteria included patients with hemorrhagic transformation on computed tomography (CT) scan, lacunar or brainstem infarct, seizure attack, or significant head trauma (defined as a Glasgow Coma Scale score of 3 to 8). Additionally, those with uncontrolled hypertension, uncorrected coagulopathy, a history of malignancy, major surgery within the past 30 days, pregnancy, HIV infection, or significant illness were also excluded.
Trial process
The trial was conducted at Taipei Medical University – Shuang Ho Hospital in Taiwan. Eligible patients were assigned to either cohort 1 or cohort 2. Baseline characteristics and vital signs were recorded before the administration of UMC119-06. Patients received a single-dose IV infusion of UMC119-06 during the treatment phase. A physical examination was performed 30 minutes after the infusion was completed. To mitigate any potential safety risks, patients were hospitalized for 14 days during the DLT evaluation period. Safety data from the first patient in each cohort, as well as from all patients in completed cohorts, were reviewed by the Dose Escalation Committee (DEC) before further patient enrollment or dose escalation. No interim efficacy analysis was planned for this trial. Clinical assessments were conducted within 3 months post-treatment, with an additional 1-year safety follow-up (Figure 1).
Outcomes assessment
The primary endpoints of the study were the safety, tolerability, and maximum feasible dose (MFD) of UMC119-06 in patients with acute ischemic stroke. Safety was monitored throughout the study via vital signs, laboratory tests, electrocardiography, physical examinations, and adverse events (AEs/TEAEs). The MFD was determined based on the occurrence of dose-limiting toxicities (DLTs) among TEAEs, with data reviewed by the Dose Escalation Committee (DEC) at each dose level.
Secondary endpoints included the degree of clinical functional improvement, assessed by changes in the modified Rankin Scale (mRS), National Institutes of Health Stroke Scale (NIHSS), Barthel Index (BI), and infarct volume on brain MRI. Among these, mRS and BI reflect the level of dependence in activities of daily living, while NIHSS evaluates the severity of neurological deficits due to stroke. Infarct volume was measured on MRI using RAPID software, with regions showing an apparent diffusion coefficient (ADC) < 620 defined as infarcted tissue.
Adverse events
According to 21 CFR 312.32(a), an adverse event (AE) is defined as any untoward sign, symptom, or disease occurring after treatment, whether reported by the patient or representing a worsening of a pre-existing condition, and not necessarily causally related to the investigational drug or intervention.
Any abnormal findings on physical examination or laboratory tests that were assessed by the investigator as clinically significant and that occurred after initiation of treatment were recorded as AEs.
Statistical analysis
The study primarily used descriptive analyses for safety data. Exploratory inferential analyses could be performed with a significance level of p = 0.05. SAS and GraphPad Prism software were used for data processing and presentation.
Results
Patient characteristics
A total of 8 patients were screened during the trial, of whom 2 did not meet the eligibility criteria, resulting in the enrollment of 6 patients. These patients were sequentially assigned to one of the two dose cohorts, with 3 patients in each cohort. One patient in cohort 1 withdrew early upon the principal investigator’s suggestion. A total of 5 patients completed the study (Figure 1). All 6 enrolled patients were included in the safety population. Of the 6 patients, 4 (66.7%) were female. The mean age was 64.0 ± 9.8 years, and the mean body weight was 64.4 ± 8.7 kilograms. All patients had a history of hypertension; 4 had diabetes mellitus, 5 had dyslipidemia, 1 had atrial fibrillation, and 2 had a prior stroke. The median NIHSS score was 9.5 (range 5-17).
Safety outcomes
UMC119-06 was evaluated at two dose levels of 1×10⁶ and 5×10⁶ cells/kg. Among the 6 patients, 83.3% experienced AEs/TEAEs and 66.7% experienced SAEs; however, no dose-limiting toxicities (DLTs) were observed. All TEAEs and SAEs were assessed as unrelated or unlikely to be related to the investigational product, and were mainly associated with infections and respiratory complications. No deaths occurred, and no significant safety concerns related to UMC119-06 were identified.
Efficacy outcomes
Clinical function assessed by mRS, NIHSS, Barthel Index (BI), and brain MRI showed a clear trend toward improvement after treatment. By the end of follow-up (day 450), most patients achieved an mRS of 0–1, BI scores of 85–100 (indicating near-complete independence), and NIHSS scores decreased by >3 points from day 30, with greater improvement observed in the high-dose group. Brain MRI demonstrated a marked reduction in infarct volume, with 5 of 6 patients reaching an infarct volume of 0 mL by day 30 and the remaining patient by day 90, which was maintained thereafter, suggesting favorable brain tissue recovery following UMC119-06 treatment.
Exploratory outcomes
Biomarkers related to acute ischemic stroke (AIS), including interleukin-1 beta (IL-1β), interleukin-6 (IL-6), interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), vascular endothelial growth factor (VEGF), and hepatocyte growth factor (HGF), were evaluated throughout the study. Among these markers, IL-1β and IFN-γ were mostly undetectable.
From day 0 to day 30, IL-6 levels increased, with a mean increase of 24.8 pg/mL, while TNF-α increased only slightly by 0.8 pg/mL over the same period. Both VEGF and HGF peaked on day 7 and then gradually declined over time. The mean change in VEGF from day 0 to day 90 increased by 50.2 pg/mL, whereas HGF levels decreased by 40.9 pg/mL over the same period.
Discussion
This phase I study showed that UMC119-06 was well tolerated, with no dose-limiting toxicities observed and no serious adverse events identified as directly related to the investigational product. Although the rates of AEs and SAEs were high, these events were largely consistent with common post-stroke complications and did not show a dose-dependent trend, thereby supporting continued clinical development of this product.
Regarding efficacy, the study observed trends toward improvement in neurological function and independence in daily activities, along with reductions in infarct volume on MRI, suggesting the neuroprotective and neurorestorative potential of UCMSCs. These findings are consistent with several previous studies of MSCs in stroke, particularly when administered in the early phase, and suggest that the cell source (UCMSCs versus autologous MSCs) may influence therapeutic outcomes.
Changes in biomarkers suggest that UMC119-06 may exert its effects through immunomodulation, promotion of angiogenesis, and tissue repair, as reflected by early increases in VEGF and HGF and alterations in IL-6 and TNF-α, indicating roles in post-ischemic inflammation and regeneration.
However, the study has several limitations, including a very small sample size, an open-label design without a control group, and incomplete control of factors influencing recovery; therefore, definitive conclusions regarding therapeutic efficacy cannot be drawn. Future studies with larger sample sizes, controlled designs, and longer follow-up are needed to clearly establish the safety, optimal dose, and true efficacy of UCMSC therapy in AIS, as well as to further elucidate the underlying biological mechanisms through biomarker analyses.
Conclusion
This phase I study indicates that UMC119-06 (derived from human umbilical cord mesenchymal stem cells, UCMSCs) is safe and well tolerated in patients with acute ischemic stroke (AIS), with signals of preliminary efficacy. However, large-scale randomized trials are required to confirm its efficacy, optimize dosing and administration, elucidate mechanisms of action through biomarker analyses, and continue close monitoring of serious adverse events before widespread clinical application.
References
Chan, L., Chen, J.H., Hong, C.T., Lin, Y.C., Chang, J.H., Chen, C.C., Hsuan, Y.C.Y., Hu, C.J. (2026). A Phase I, Open-Label, Dose-Escalation Study to Evaluate the Safety and Tolerability of Intravenous UMC119-06 in Patients with Acute Ischemic Stroke. International Journal of Medical Sciences, 23(2), 576-584.
Source: Int J Med Sci